Perspective: Exploring The Relationship Between Climate Change And Dementia
- Jenna Rector
- Jun 12
- 20 min read

Abstract
Alzheimer’s Disease is a neurodegenerative disorder in which many pathways and processes are characterized by decreased or ablated function. Researchers have been exploring potential causes and effects of Alzheimer's on the brain, but little is understood about the role of climate change in its pathology. Climate change has long been linked to a significant increase in global temperature, but studies showing evidence of a relationship between increasing temperature and an elevated risk of developing neurodegenerative disorders and age-related deficits have only recently emerged. Because the effects of climate change are just now being understood and more is being uncovered in the field of neurodegenerative diseases every day, there is limited research on the combination of these studies. This multidisciplinary perspective aims to explain our current understanding of climate change and neurological deficits, with a heavy emphasis on Alzheimer's. By exploring the impacts of chronic and acute heat stress on three major neurological mechanisms, I explore the biochemical basis of Alzheimer's in the face of climate change and offer insights to potential new targets for AD therapeutics.
Introduction
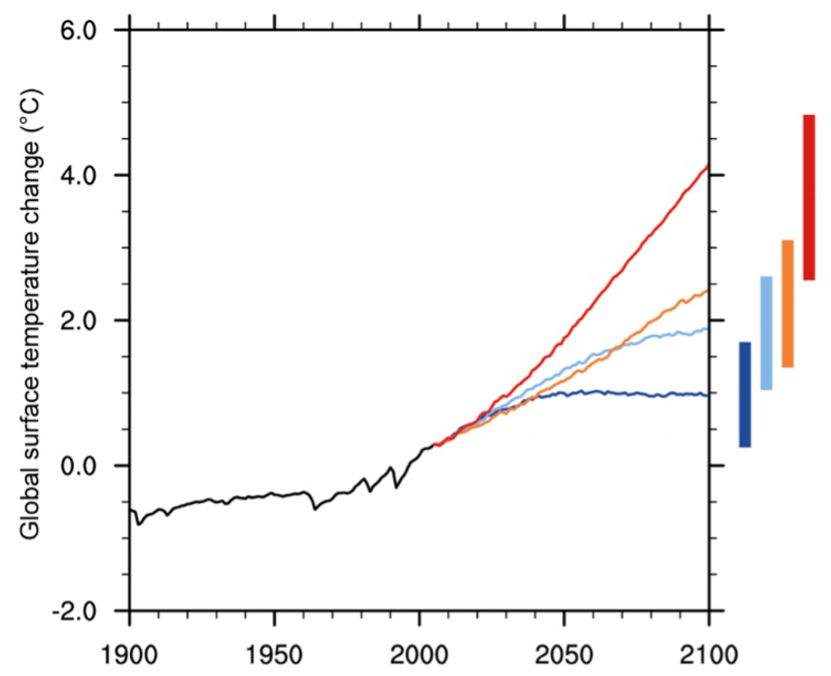
According to global records, 2023 was the warmest year in history, hitting 1.18°C (2.12°F) above the 20th century average. This is evidence of a significant increase in global average temperature (Lindsey & Dahlman, 2020). Further, climate change projections by the Environmental Protection Agency (EPA) predict an increase in the average global temperature as high as 8.6°F, as shown in Figure 1 (US EPA).
While many factors have led to the evident temperature increase, this report is concerned with the role it plays in human health, specifically in ageing and risk of developing Alzheimer’s Disease (AD). First, we will explore ageing, longevity, and dementia as it relates to climate change, where climate change will be represented by temperature variations or chronic exposure to heat. Next, we aim to understand the effects of climate change, in terms of acute exposure to heat, so-called “heat stress”, on the biochemical mechanisms involved in dementia, specifically Alzheimer’s disease. Finally, we will then discuss how this information relates to Alzheimer’s treatment options and I will provide my concluding remarks.
While several studies have linked exposure to elevated temperatures or temperature variations to increased mortality rates (Shi et al., 2015, 2016), the relationship between long-term exposure to temperature variations and risk of dementia was recently explored. A 2019 study aimed to understand the correlation between temperature fluctuations and the number of dementia-related hospitalizations in New England. The results suggested an increased risk of dementia-related hospital admissions with an increase in prolonged exposure to seasonal temperature variations. This result was consistent across all populations. Conclusions were drawn suggesting that age-related decline of thermoregulation processes makes older adults more prone to issues in adapting to body temperature changes and, therefore, more susceptible to thermoregulatory disorders (Wei et al., 2019). In light of this finding, it is important to understand how temperature changes affect the central nervous system at a molecular level, leading to its apparent influence on the risk of developing dementia.
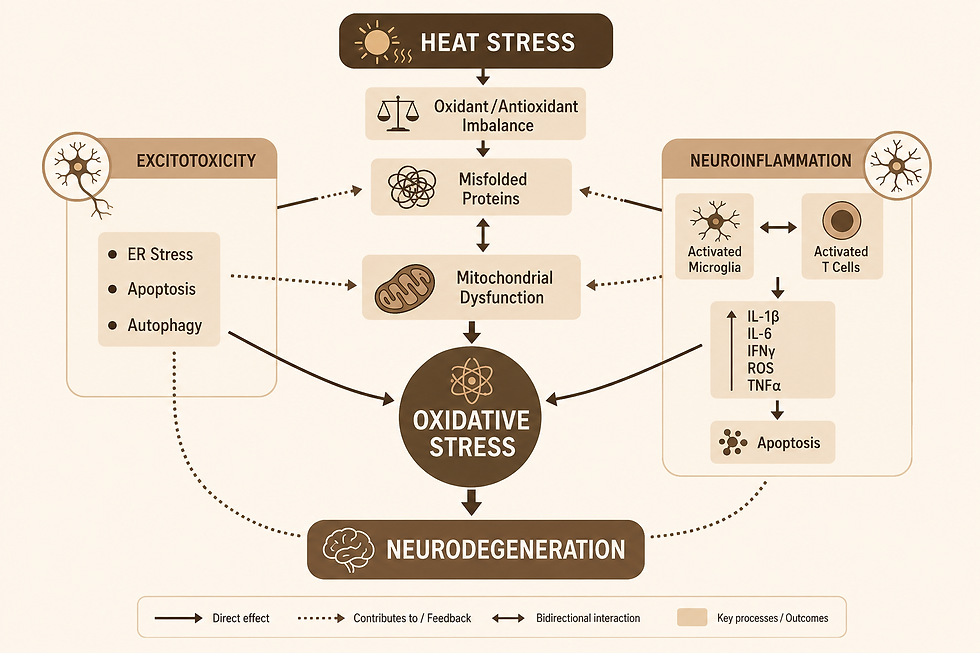
Like many other neurodegenerative diseases, AD pathology is not completely understood. The study of AD involves a complex web of various neurobiological pathways which would normally work together to maintain homeostasis and support healthy cognitive function. However, AD is hallmarked by characteristic β-amyloid plaques and tau proteins that disrupt this healthy function. While some mysteries still remain, research has advanced in understanding the AD mechanism of action. It is currently understood that the dysfunction of β-amyloid and tau proteins results in a cascade of downstream effects such as neuroinflammation, oxidative stress, and excitotoxicity, which ultimately lead to cell death and neurodegeneration. In this perspective, the underlying biochemical mechanisms of AD pathology are divided into these three categories: (1) neuroinflammation, (2) oxidative stress, and (3) excitotoxicity.
Neuroinflammtion: Evidence shows that the aggregation of β-amyloid plaques can trigger the activation of microglial cells, which promote neuroinflammation as an immune response (Kiraly, Foss, & Giordano, 2023). Chronic neuroinflammation eventually leads to cell death and neurodegeneration.
Oxidative stress: The severity of oxidative stress has come to light somewhat recently. Its onset begins when β-amyloid aggregates interact with chemical species to form free radicals. Because these radicals are inherently unstable, they quickly react with DNA, lipids and important proteins. The modification of these species disrupts their structural integrity and functionality, leading to the dysfunction of the neural pathways and detrimental cognitive effects (Perluigi, Domenico, & Butterfield, 2024).
Excitotoxicity: The most notable source of excitotoxicity is prolonged depolarization. When a stimulus reaches the dendrite of a neuron, it sends an electrical pulse down the neuron's axon through a process called depolarization. At the axon terminus, the electrical signal is converted to a chemical signal, during which voltage-gated calcium channels open and glutamate is released into the synapse. When this process is disrupted in an AD brain, glutamate - an excitatory neurotransmitter - builds up in the synapse, resulting in cell death through apoptosis.
Aside from a pivotal role in AD pathology, these processes share another common feature: temperature dependence. Understanding their specific sensitivity to temperature helps us understand heat-induced stress, which exacerbates dysfunction and could lead to accelerated AD progression, as shown in Figure 2 (Bongioanni et al., 2021). Thus, we can draw a connection between climate change and AD progression through heat-induced pathway disruption. This perspective aims to review our current understanding of these underlying AD mechanisms and explore how climate change could accelerate disease progression through thermal stress.
The Effect of Thermal Stress on the Brain
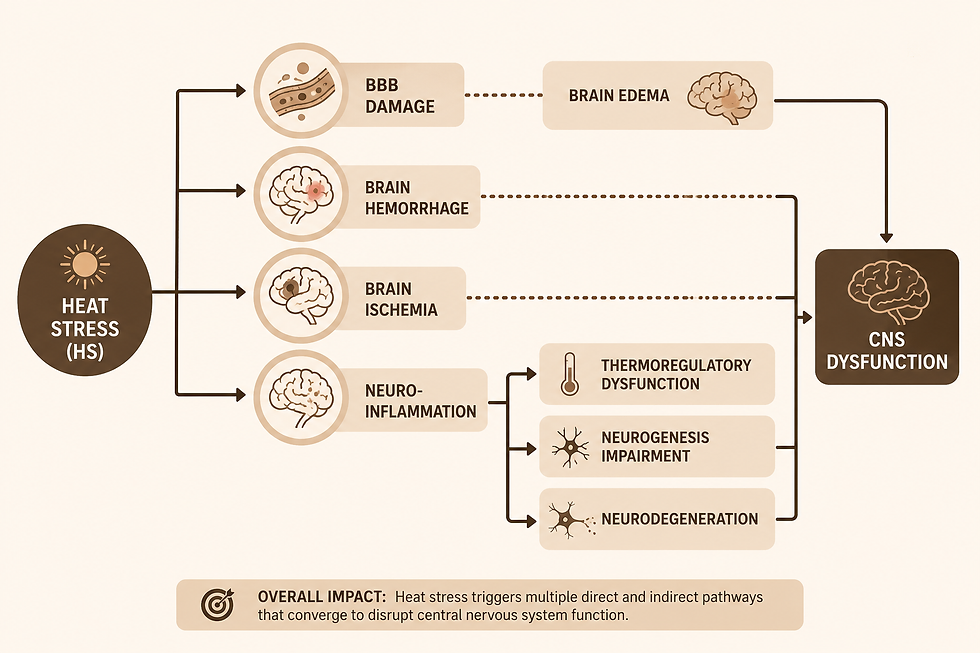
While researchers have previously studied the influence of high temperatures on aging and dementia, we have only recently begun to understand the impact of thermal stress on the nervous system. Evidence has been shown that heat stress negatively affects the central nervous system (CNS) structure and function (Walter & Carraretto, 2016), as shown in Figure 3. Other studies expand on this, correlating heat stress with reduction in brain perfusion, CNS fatigue, destruction of the blood-brain barrier, and brain edema, leading to modifications in neuronal circuits, neurological defects, and even brain atrophy (Sharma et al. 1998; Nybo et al., 2007). Recurring brain injuries, including hemorrhage and ischemia, have also been studied in terms of induced thermal stress Zhang & Li, 2014; Li et al., 2015). However, perhaps the most threatening and severe consequence of heat stress is neuroinflammation (Medzhitov, 2008; Lee et al., 2015; Chauhan et al., 2017; Zhao et al., 2021a,b). Over the last decade, extensive research has associated chronic neuroinflammation with synaptic dysfunction, inhibition of neurogenesis, neuronal death, and cognitive impairment (Wang et al., 2023). These findings suggest that thermal stress can induce responses in neurological mechanisms that accelerate AD pathology.
A 2015 study explored the relationship between thermal stress and memory impairment in mice. The researchers used a longitudinal study involving mice exposed to higher-than-normal temperatures over 7, 14, or 42 days. Then, they examined cognitive behavior through tests such as the Y-maze, passive avoidance, and the novel subject recognition test, and analyzed the physiological responses in the hippocampus. They found that pathophysiological responses to heat stress were associated with neuroinflammation in the hippocampus, leading to impaired cognitive ability (Lee et al., 2015).
The behavior tests revealed lowered cognitive ability in the heat-exposed mice compared to control mice. While the results of the Y-maze did not change upon prolonged exposure to heat, the results of the passive avoidance test showed that retention time of the heat-exposed group significantly declined. Additionally, in the novel object experiment, the heat-exposed mice spent about the same time exploring the novel object and familiar objects, while the control mice spent significantly more time exploring the novel object compared to the familiar one. Together, the results of the passive avoidance and novel recognition tests show that heat stress inhibits cognitive function in mice in a time-dependent manner (Lee et al., 2015).
The behavioral data from this experiment suggest heat-induced hippocampal memory impairment (Lee et al., 2015). The Y-maze is designed to test procedural and motor-control memory (i.e. “muscle memory”), which is regulated by the cerebellum. Thus, it is reasonable that thermal stress did not affect the results of this test. Instead, it appeared to cause dysfunction of the hippocampus, which is involved in learning and the formation of new memories, shown by the results of the passive avoidance and novel object experiments.
Analysis of hippocampal tissue from the heat-exposed mice in the 2015 study reflect this hypothesis. Neuroinflammation in the hippocampus was recognized by activation of specific proteins. Western blot confirmed elevated levels of heat shock protein 70 (Hsp70) and a stress-response protein (c-fos), as well as the activation of nuclear factor-kappa B (NF-κB), a standard regulator of neuroinflammation. Immunohistochemistry also confirmed an elevated number of astrocytes following heat exposure (Lee et al., 2015).
A more recent study in 2023 also reported an elevated number of astrocytes and glia, increased expression of Hsp70, and the activation of the NF-κB pathway associated with heat stress-induced neuroinflammation (Wang et al., 2023). Researchers went on to explore the underlying biological mechanism of neuroinflammation regarding these specific proteins. Hsps are synthesized by glia in response to heat stress, and they act as molecular chaperones to promote proper folding of polypeptides and repair damaged proteins (Taylor et al., 2007). Thus, their role in maintaining protein homeostasis is pivotal. Hsp70 is a specific Hsp synthesized by oligodendrocytes, rather than glia, that initiates neuroinflammation upon binding to its receptor, Toll-like receptor 4 (TLR4), expressed on the membrane of microglia and astrocytes. This binding event triggers a signaling pathway during which downstream targets are recruited, and eventually NF-κB is activated. The activation of NF-κB recruits pro-inflammatory mediators that accelerate the development of neuroinflammation (Wang et al., 2023). Thus, the increased expression of Hsp70 and activation of the NF-κB pathway not only demonstrates the development of neuroinflammation, but it also highlights a negative feedback mechanism in which the neuroinflammation is highly regulated and accelerated.
In addition to proteins associated with the NF-κB pathway, prolonged exposure to heat has also been linked to the upregulated gene expression of proinflammatory IL-1β, IL-6, TNF-α, and nitric oxide synthase (NOS) with subsequent depletion of neuronal and synaptic density in the hippocampus (Lee et al., 2015, Christoforidou et al., 2020). Specifically, TNF-α is associated with cell death through the extrinsic apoptotic pathway, in which death receptors are activated. Activation of death receptors, FADD and TRADD, by cytokines, Fas and TNF- α, respectively, causes a cascade of caspase activation (namely caspase 8 and caspase 3), resulting in cell death.
As shown above, heat stress can induce the release of proinflammatory proteins by microglia. Some proteins, including IL-1β, IL-6, TNF- α, NOS, and reactive oxygen species (ROS), not only further promote neuroinflammation, but also negatively impact AD progression in other ways (Bongioanni et al., 2021). Protein mutations, oxidative stress, impairment of protein homeostasis, and DNA modifications are all significant markers of AD pathology. Of note, oxidative stress is a relatively novel phenomenon based on an imbalance between the production and accumulation of ROS and the ability of a biological system to remove these very reactive, toxic species (Pizzino et al., 2017). When produced in excess, ROS spread throughout the brain causing damage to various systems and proteins, thereby oxidatively modifying them and usually rendering them dysfunctional. Once oxidative stress is established in the brain, it progresses quickly and without prejudice, making it extremely detrimental to overall brain health and a biomarker of several neurodegenerative diseases, including AD.
Various oxidatively modified, or dysfunctional, processes can work together to initiate the formation and deposition of protein aggregates. Not only do these aggregates activate an inflammatory response, thereby exacerbating neuroinflammation, but they also can overwhelm the microglial clearance systems causing an accumulation of Aβ plaques, neurofibrillary tau tangles, apoptotic bodies, and other debris. When the clearance systems are overwhelmed, this only accelerates damage via protein aggregates and debris that cannot be effectively removed from the brain. Here, I note another dangerous negative feedback mechanism which is a recurring theme across all AD pathogenic pathways discussed in this review. It has been reported that oxidative stress appears early in the clinical stages of AD, before the onset of dementia (Perluigi, Domenico, & Butterfield, 2024). Perhaps this is due to the intrinsic regulation of ROS production and subsequent oxidative modification. This is probably why clinical symptoms of AD gradually worsen from the onset of pre-clinical AD to mild cognitive impairment (MCI) up until severe dementia.
Proinflammatory responses are coordinated by the mitochondria (Andrieux et al., 2021). As heat stress has been known to induce mitochondrial dysfunction (Akbarian et al., 2016), a reduced ability to balance oxidants and antioxidants has been observed in heat stress-induced models. This imbalance can lead to the overproduction of ROS and the accumulation of free radicals, promoting oxidative stress in neurons (Bongioanni et al., 2021). A 2007 study linked heat stress to elevated levels of the superoxide anion (Mujahid et al., 2007), a powerful free radical with potentially devastating neurochemical effects. Further, a 2011 study contributed the reduced expression of the antioxidant superoxide dehydrogenase (SOD) to high temperature exposure (El Orabi et al., 2011). The overproduction of superoxide anion (O2-.), without the balance of SOD, can lead to serious downstream consequences, including protein oxidation and lipid peroxidation. The oxidant-antioxidant imbalance also leads to the production of other free radicals (Swomley et al., 2014), including nitrous oxide (NO), which induces protein nitration. The combination of lipid peroxidation, protein oxidation, and protein nitration have serious structural and functional consequences of downstream proteins. More information and figures can be found in this publication. The production of ROS also involves NADPH oxidases (NOX) (Moon et al., 2010; Kikusato et al., 2015), which leads to dysfunction in the electron transport chain, causing further increased levels of superoxide and other ROS. Thus, the production of ROS and generation of free radicals are an important basis of oxidative damage.
This cycle also has important downstream effects, including lipid peroxidation, protein and DNA modification/oxidation, and mitochondrial membrane permeabilization, leading to the release of proapoptotic factors and subsequent cell death. Similar to protein oxidation, lipid peroxidation also involves an attack by oxidant free radicals. The free radicals target a partially positive carbon of a polyunsaturated fatty acid and pick up an allylic hydrogen to occupy the unpaired electron on the radical. Now, the lipid radical can react with oxygen to produce a lipid peroxyl radical. Then, this radical will react with another allylic hydrogen to regenerate a lipid radical and form a lipid hydroperoxide, from which a severely detrimental product, HNE, is formed. HNE will go on to disturb many downstream processes, and the lipid radical will continue to participate in lipid peroxidation chain mechanism until there are no more available substrates.
The endoplasmic reticulum (ER) plays a role in lipid metabolism, Ca2+ transport, and autophagy through the formation of mitochondria-associated ER membranes (MAMs) (Malhotra & Kaufman, 2011); thus, the oxidative modification of ER and mitochondrial dysfunction may underlie the mechanism of heat stress-induced neurodegeneration. The association of mitochondria and ER has been studied somewhat extensively in terms of oxidative stress. The relationship is thought to play a role in overall cerebral oxidative stress and Tauopathies with heat stress as a pro-oxidant factor (Slimen et al., 2014; Chauderlier et al., 2017; Chauhan et al., 2017).
Excitotoxicity refers to an excess amount of receptor activation by an excitatory neurotransmitter (usually glutamate). One way excitotoxicity can occur is from overall increased brain excitability, which usually involves a constant release of an excitatory neurotransmitter, subsequently leading to constant neuron depolarization. This is especially detrimental when coupled with lower levels of the counteracting inhibitory neurotransmitters (i.e. GABA). One study showed that heat stress in rats can increase levels of excitatory neurotransmitters (glutamate and aspartate) overall, while decreasing the concentrations of inhibitory neurotransmitters (GABA and glycine). Depressed levels of inhibitory neurotransmitter was especially related to reduced GABAergic synaptic transmission in the hippocampus. This shift toward excitatory neurotransmission enhanced neurodegeneration via excitotoxicity, especially in the hippocampus (Sharma, 2006; Qu et al., 2007). An extended study further demonstrated that increasing the temperature associated with heat stress (i.e. prolonged hyperthermia) led to even higher glutamate concentrations circulating throughout the whole brain (Zlotnik et al., 2010). Later in 2012, another experiment showed that hyperthermia-related excitotoxicity resulted in an increased activity of hippocampal pyramidal cells (Kim & Connors, 2012), which are involved in spatial memory and encoding contextual and emotional information (Graves et al., 2012).
Another method of excitotoxicity stems from a lack of synaptic glutamate removal, leaving high concentrations of glutamate in the synapse that continue to activate glutamate receptors and depolarize the cell. This can occur via two ways: (1) re-uptake, where glutamate is transported directly back into either the presynaptic terminal via high-affinity glutamate transporters (i.e. EAAT2) or into the post-synaptic terminal via EAAT3/4, and (2) uptake through the glutamine shuttle. The glutamine shuttle, transports glutamate between astrocytes and the presynaptic terminal (Brady & Siegel, 2011). First, glutamate is taken up into astrocytes via EAAT2, where it is then converted to glutamine. Glutamine is transported out of the astrocyte and into the presynaptic neuron through SN and SA transporters, respectively. Then, the mitochondria convert glutamine back into glutamate using phosphate-activated glutaminase, and glutamate is again ready to participate in synaptic transmission. Again, we see that mitochondria and glial cells play a pivotal role in a pathway affected by AD pathology. Oxidative modification of any of these key players, especially glutamate transporters, can lead to excitotoxicity and consequential cell death.
Excitotoxicity induces cell death mainly through calcium overload and the intrinsic apoptotic pathway. In this mechanism, several mitochondrial changes occur upon the increased uptake up Ca2+, including an increased production of ROS and an increase in membrane permeability due to the opening of the mitochondrial permeability transition pore (MPTP). Upon these changes, cytochrome C, then subsequently apoptosis-inducing factor (AIF) are released. Finally, a cascade of caspases (namely caspase 9 and caspase 3) is activated resulting in cell death (Fhearraigh, 2024). Thus, an interplay between AD pathogenic pathways and critical proteins involved can again be observed in the complex process of neurodegeneration.
Moving forward with AD therapeutics in the face of climate change
Climate models use data and computer programs to produce projections about how the earth’s climate may change over time. The graph in Figure 1 was made using data from such models. As scientists and activists continue to raise awareness around climate change, more research has come to light. This data is useful in making estimations about the future state of our planet, and I predict that, eventually, we can use these projections to target geographical areas especially susceptible to certain diseases.
So far, this perspective has shown evidence of a connection between an elevated risk of AD and climate change through the lens of heat stress. This poses a question for future directions in both fields: Can we use climate change projections to identify areas that are susceptible to significant temperature increases and use this information to target these areas for AD therapeutics? Further, can we combine these projections with socioeconomic data to diminish environmental and clinical injustice?
There is a large amount of funding dedicated to research in and out of the clinical setting for AD, and much progress has been made. However, the potential for accelerated progression of AD by thermal stress may decrease the positive impacts of these treatments. For example, increasing antioxidant levels in the brain is a common method to slow oxidative stress. However, with the accelerated rate of oxidant and free radical production under heat stress, the administration of antioxidants may no longer be effective. Because we are in somewhat uncharted territory, we have little knowledge about how AD (and other neurodegenerative diseases) will progress in the face of climate change. Thus, it is important to note that the limitations of current therapeutic approaches may be associated with the progression of climate change.
Although it is easy to be discouraged by the current limitations, this perspective offers a new direction. Using this newly established correlation between AD and climate change, thermal stress mechanisms shine light on potential new targets for AD through new pathways and thermoregulatory systems. Heat-induced stress has provided new insights to AD pathology in terms of neuroinflammation, oxidative stress, and excitotoxicity. Could these new insights lead to new therapeutic targets for AD? Understanding the molecular and biochemical basis of AD under heat stress has highlighted the importance of several pathways and processes, including mitochondrial and ER dysfunction, intrinsic and extrinsic apoptotic pathways, glutamate uptake, production of ROS and decreased levels of antioxidants, and many others. While some therapeutics already target some of these pathways, I predict that this new information will be useful in fine-tuning pharmacological solutions to these dysfunctional systems. I think mitochondrial dysfunction is an especially important target for AD treatments, since this review places it in nearly every AD pathogenic pathway. An overview of the role of mitochondrial dysfunction in AD and its mechanism of calcium signaling and cell death is shown in this publication (Bhatia et al., 2022). Additionally, understanding thermoregulatory pathways and developing treatments that increase their stability and functionality may offer new insights in the pharmaceutical industry. Current approaches, combined with the new evidence of heat stress and research of thermoregulation could provide hope for potential treatments for AD.
Conclusions
This perspective serves as a review of our current understanding of AD pathogenic pathways and how they relate to climate change-induced heat stress. Based on the level of temperature sensitivity, AD pathology was divided into three major categories: (1) neuroinflammation, (2) oxidative stress, and (3) excitotoxicity. It is interesting to note that neuroinflammation is heavily localized within the hippocampus while oxidative stress and excitotoxicity spread throughout the brain. Perhaps this is due to the vast supply of oxygen and neurotransmitters throughout the brain and the mobility of free radicals.
Neuroinflammation contributes to AD pathology by microglial activation and release of proinflammatory molecules. Under acute heat stress, one such molecule, Hsp70, activates the NF-κB pathway, which recruits more proinflammatory proteins to intrinsically regulate the neuroinflammatory stress response. TNF-α and NOS are other proteins to note. TNF-α later binds to the TRADD death receptor, triggering apoptosis via the extrinsic pathway. NOS converts arginine to the nitrous oxide radical, which initiates protein nitration and causes detrimental neurological effects alone and combined with oxidative stress.
Oxidative stress refers to an imbalance between oxidants and antioxidants. This imbalance is worsened by exposure to heat stress. The resulting shift to elevated oxidant levels and depressed antioxidant levels leads to protein nitration, lipid peroxidation, and oxidative modification of DNA and proteins, rendering many neurobiological pathways dysfunctional. The overall result is neuron death and subsequent neurodegeneration, followed by clinical symptoms revolving around cognitive decline.
Excitotoxicity was the third and final topic of our discussion. In AD brain, it is understood that excitotoxicity is mostly a consequence of oxidatively modified glutamate transports (namely EAAT1/2), that disrupts the uptake of glutamate from the synapse. The high concentration of glutamate in the synaptic cleft triggers postsynaptic depolarization, leading to an overwhelming influx of Ca2+, which prompts the opening of MPTP and apoptosis via the intrinsic pathway. Heat stress models show that excitatory neurotransmitter levels are increased throughout the brain, coupled with reduced inhibitory neurotransmitter concentrations.
This network of biochemical mechanisms underlying AD is profoundly affected by heat stress, indicating that climate change - through the lens of acute and chronic heat exposure - can contribute to the onset and progression of AD. Thus, this perspective has established an interesting link between climate change and an accelerated rate or increased risk of AD. Along with climate change projection models, these novel findings may provide fresh insights for potential AD therapeutics and help us overcome obstacles in both fields. Information regarding highly susceptible geographical areas and socioeconomic classes can enable us to fine-tune our approach to treating AD and tackling environmental injustice.
References
Lindsey, R., & Dahlman, L. (2020). Climate change: Global temperature. Climate. gov, 16.
US EPA. Future of Climate Change | Climate Change Science | US EPA. (n.d.). https://19january2017snapshot.epa.gov/climate-change-science/future-climate-change_.html
Shi, L., Kloog, I., Zanobetti, A., Liu, P., & Schwartz, J. D. (2015). Impacts of temperature and its variability on mortality in New England. Nature climate change, 5(11), 988-991.
Shi, L., Liu, P., Wang, Y., Zanobetti, A., Kosheleva, A., Koutrakis, P., & Schwartz, J. (2016). Chronic effects of temperature on mortality in the Southeastern USA using satellite-based exposure metrics. Scientific Reports, 6(1), 30161.
Wei, Y., Wang, Y., Lin, C. K., Yin, K., Yang, J., Shi, L., ... & Schwartz, J. D. (2019). Associations between seasonal temperature and dementia-associated hospitalizations in New England. Environment international, 126, 228-233.
Lee, W., Moon, M., Kim, H. G., Lee, T. H., & Oh, M. S. (2015). Heat stress-induced memory impairment is associated with neuroinflammation in mice. Journal of neuroinflammation, 12, 1-13.
Wang, S., Hou, K., Gui, S., Ma, Y., Wang, S., Zhao, S., & Zhu, X. (2023). Insulin-like growth factor 1 in heat stress-induced neuroinflammation: novel perspective about the neuroprotective role of chromium. Stress Biology, 3(1), 23.
Malhotra, J. D., & Kaufman, R. J. (2011). ER stress and its functional link to mitochondria: role in cell survival and death. Cold Spring Harbor perspectives in biology, 3(9), a004424.
Graves, A. R., Moore, S. J., Bloss, E. B., Mensh, B. D., Kath, W. L., & Spruston, N. (2012). Hippocampal pyramidal neurons comprise two distinct cell types that are countermodulated by metabotropic receptors. Neuron, 76(4), 776-789.
Walter, E. J., & Carraretto, M. (2016). The neurological and cognitive consequences of hyperthermia. Critical Care, 20, 1-8.
Nybo, L. (2007). Exercise and heat stress: cerebral challenges and consequences. Progress in brain research, 162, 29-43.
Sharma, H. S., Westman, J., & Nyberg, F. (1998). Pathophysiology of brain edema and cell changes following hyperthermic brain injury. Progress in brain research, 115, 351-412.
Zhang, X. Y., & Li, J. (2014). Susceptibility-weighted imaging in heat stroke. PLoS One, 9(8), e105247.
Li, J., Zhang, X. Y., Wang, B., Zou, Z. M., Wang, P. Y., Xia, J. K., & Li, H. F. (2015). Diffusion tensor imaging of the cerebellum in patients after heat stroke. Acta Neurologica Belgica, 115, 147-150.
Lee, W., Moon, M., Kim, H. G., Lee, T. H., & Oh, M. S. (2015). Heat stress-induced memory impairment is associated with neuroinflammation in mice. Journal of neuroinflammation, 12, 1-13.
Medzhitov, R. (2008). Origin and physiological roles of inflammation. Nature, 454(7203), 428-435.
Chauhan, N. R., Kapoor, M., Singh, L. P., Gupta, R. K., Meena, R. C., Tulsawani, R., ... & Singh, S. B. (2017). Heat stress-induced neuroinflammation and aberration in monoamine levels in hypothalamus are associated with temperature dysregulation. Neuroscience, 358, 79-92.
Zhao, T., Zhu, Y., Yao, L., Liu, L., & Li, N. (2021). IGF-1 alleviates CCL4-induced hepatic cirrhosis and dysfunction of intestinal barrier through inhibition TLR4/NF-κB signaling mediated by down-regulation HMGB1. Annals of Hepatology, 26, 100560.
Zhao, Y., Zhuang, Y., Shi, Y., Xu, Z., Zhou, C., Guo, L., ... & Xu, L. (2021). Effects of N-acetyl-l-cysteine on heat stress-induced oxidative stress and inflammation in the hypothalamus of hens. Journal of Thermal Biology, 98, 102927.
Taylor, A. R., Robinson, M. B., Gifondorwa, D. J., Tytell, M., & Milligan, C. E. (2007). Regulation of heat shock protein 70 release in astrocytes: role of signaling kinases. Developmental neurobiology, 67(13), 1815-1829.
Christoforidou, E., et al., 2020. Potential of activated microglia as a source of dysregulated extracellular microRNAs contributing to neurodegeneration in amyotrophic lateral sclerosis. J. Neuroinflammation 17, 135.
Brady, S., & Siegel, G. (2011). Basic neurochemistry: principles of molecular, cellular, and medical neurobiology. Academic press.
Bongioanni, P., Del Carratore, R., Corbianco, S., Diana, A., Cavallini, G., Masciandaro, S. M., ... & Buizza, R. (2021). Climate change and neurodegenerative diseases. Environmental Research, 201, 111511.
Perluigi, M., Di Domenico, F., & Butterfield, D. A. (2024). Oxidative damage in neurodegeneration: Roles in the pathogenesis and progression of Alzheimer disease. Physiological Reviews, 104(1), 103-197.
Akbarian, A., et al., 2016. Association between heat stress and oxidative stress in poultry; mitochondrial dysfunction and dietary interventions with phytochemicals. J. Anim. Sci. Biotechnol. 7, 37–50.
Mujahid, A., et al., 2007. Mitochondrial oxidative damage in chicken skeletal muscle induced by acute heat stress. J. Poultry Sci. 44, 439–444.
Kikusato, M., Yoshida, H., Furukawa, K., & Toyomizu, M. (2015). Effect of heat stress-induced production of mitochondrial reactive oxygen species on NADPH oxidase and heme oxygenase-1 mRNA levels in avian muscle cells. Journal of thermal biology, 52, 8-13.
Moon, E. J., Sonveaux, P., Porporato, P. E., Danhier, P., Gallez, B., Batinic-Haberle, I., ... & Dewhirst, M. W. (2010). NADPH oxidase-mediated reactive oxygen species production activates hypoxia-inducible factor-1 (HIF-1) via the ERK pathway after hyperthermia treatment. Proceedings of the National Academy of Sciences, 107(47), 20477-20482.
El-Orabi, N., et al., 2011. Heat-induced inhibition of superoxide dismutase and accumulation of reactive oxygen species leads to HT22 neuronal cell death. J. Therm. Biol. 36, 49–56.
Chauderlier, A., et al., 2017. In vivo hyperthermic stress model: an easy tool to study the effects of oxidative stress on neuronal Tau functionality in mouse brain. Methods Mol. Biol. 1523, 369–373.
Chauhan, N.R., et al., 2017. Heat stress-induced neuroinflammation and aberration in monoamine levels in hypothalamus are associated with temperature dysregulation. Neuroscience 358, 79–92.
Slimen, I.B., et al., 2014. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A Rev. Int. J. Hyperth. 30, 513–523.
Sharma, H.S., 2006. Hyperthermia influences excitatory and inhibitory amino acid neurotransmitters in the central nervous system. An experimental study in the rat using behavioural, biochemical, pharmacological, and morphological approaches. J. Neural. Transm. 113, 497–519DOI.
Qu, L., et al., 2007. Hyperthermia decreases GABAergic synaptic transmission in hippocampal neurons of immature rats. Neurobiol. Dis. 27, 320–327.
Zlotnik, A., et al., 2010. The effect of hyperthermia on blood glutamate levels. Anesth. Analg. 111, 1497–1504.
Kim, J.A., Connors, B.W., 2012. High temperatures alter physiological properties of pyramidal cells and inhibitory interneurons in hippocampus. Front. Cell. Neurosci. 6, 27.
Bhatia, S., Rawal, R., Sharma, P., Singh, T., Singh, M., & Singh, V. (2022). Mitochondrial dysfunction in Alzheimer’s disease: opportunities for drug development. Current Neuropharmacology, 20(4), 675.
Pizzino, G., Irrera, N., Cucinotta, M., Pallio, G., Mannino, F., Arcoraci, V., ... & Bitto, A. (2017). Oxidative stress: harms and benefits for human health. Oxidative medicine and cellular longevity, 2017.
Andrieux, P., Chevillard, C., Cunha-Neto, E., & Nunes, J. P. S. (2021). Mitochondria as a cellular hub in infection and inflammation. International journal of molecular sciences, 22(21), 11338.
Fhearraigh, S. M. (2024). Apoptosis (intrinsic & extrinsic pathways). Assay Genie. https://www.assaygenie.com/apoptosis-intrinsic-extrinsic-cell-death-pathways
Kiraly, M., Foss, J. F., & Giordano, T. (2023). Neuroinflammation, Its Role in Alzheimer’s Disease and Therapeutic Strategies. The Journal of Prevention of Alzheimer's Disease, 10(4), 686-698.
Swomley, A. M., Förster, S., Keeney, J. T., Triplett, J., Zhang, Z., Sultana, R., & Butterfield, D. A. (2014). Abeta, oxidative stress in Alzheimer disease: evidence based on proteomics studies. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease, 1842(8), 1248-1257.

Comments